 中文网站
中文网站
Tidligt om morgenen den 29. december offentliggjorde NEJM online et nyt klinisk fase III-studie af den nye kinesiske coronavirus VV116. Resultaterne viste, at VV116 ikke var værre end Paxlovid (nematovir/ritonavir) med hensyn til varighed af klinisk bedring og havde færre bivirkninger.

Billedkilde: NEJM
Median restitutionstid 4 dage, bivirkningsrate 67,4 %
VV116 er et oralt nukleosid-anti-nyt coronavirus (SARS-CoV-2) lægemiddel udviklet i samarbejde med Junsit og Wang Shan Wang Shui, og er en RdRp-hæmmer sammen med Gileads remdesivir, Merck Sharp & Dohmes molnupiravir og Real Biologics' azelvudin.
I 2021 blev et fase II klinisk forsøg med VV116 afsluttet i Usbekistan. Resultaterne af undersøgelsen viste, at VV116-gruppen bedre kunne forbedre kliniske symptomer og reducere risikoen for progression til den kritiske form og død signifikant sammenlignet med kontrolgruppen. Baseret på de positive resultater af dette forsøg er VV116 blevet godkendt i Usbekistan til behandling af patienter med moderat til svær COVID-19 og er blevet det første nye orale koronarlægemiddel, der er godkendt til markedsføring i udlandet i Kina [1].
Dette fase III kliniske forsøg[2] (NCT05341609), ledet af professor Zhao Ren fra Shanghai Ruijin Hospital, professor Gaoyuan fra Shanghai Renji Hospital og akademiker Ning Guang fra Shanghai Ruijin Hospital, blev afsluttet under udbruddet forårsaget af Omicron-varianten (B.1.1.529) fra marts til maj i Shanghai med det formål at evaluere effekten og sikkerheden af VV116 versus Paxlovid til tidlig behandling af patienter med mild til moderat COVID-19. Formålet var at evaluere effekten og sikkerheden af VV116 versus Paxlovid til tidlig behandling af patienter med mild til moderat COVID-19.

Billedkilde: Reference 2
Et multicenter, observatørblindet, randomiseret, kontrolleret forsøg med 822 voksne Covid-19-patienter med høj risiko for progression og med milde til moderate symptomer blev udført mellem 4. april og 2. maj 2022 for at vurdere deltagernes egnethed fra syv hospitaler i Shanghai, Kina. I sidste ende modtog 771 deltagere enten VV116 (384 mg, 600 mg hver 12. time på dag 1 og 300 mg hver 12. time på dag 2-5) eller Paxovid (387 mg, 300 mg nimatuvir + 100 mg ritonavir hver 12. time i 5 dage) som oral medicin.
Resultaterne af dette kliniske studie viste, at tidlig behandling med VV116 for mild til moderat COVID-19 opfyldte det primære endepunkt (tid til vedvarende klinisk bedring) forudsagt af den kliniske protokol: mediantiden til klinisk bedring var 4 dage i VV116-gruppen og 5 dage i Paxlovid-gruppen (hazard ratio, 1,17; 95% CI, 1,02 til 1,36; nedre grænse >0,8).
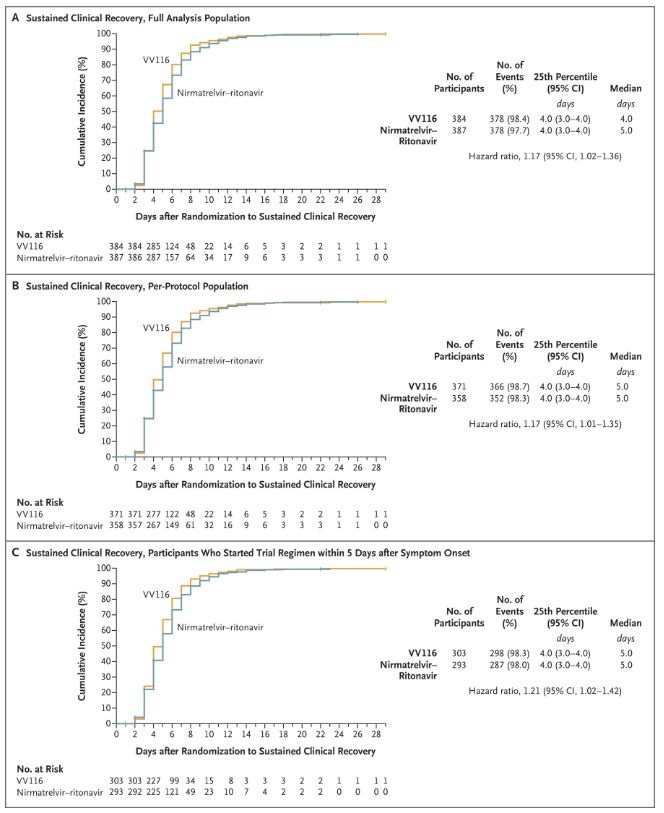
Opretholdelse af klinisk restitutionstid
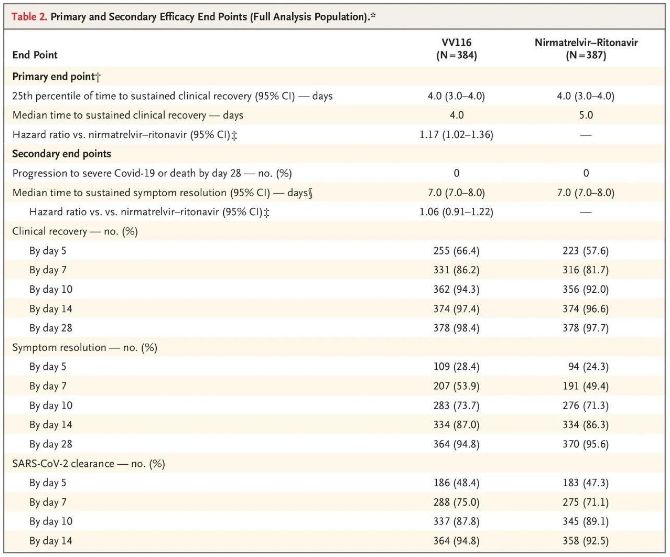
Primære og sekundære effektmål (omfattende analyse af populationen)
Billedkilde: Reference 2
Med hensyn til sikkerhed rapporterede deltagerne, der fik VV116, færre bivirkninger (67,4%) end dem, der fik Paxlovid (77,3%) ved 28-dages opfølgning, og forekomsten af grad 3/4 bivirkninger var lavere for VV116 (2,6%) end for Paxlovid (5,7%).
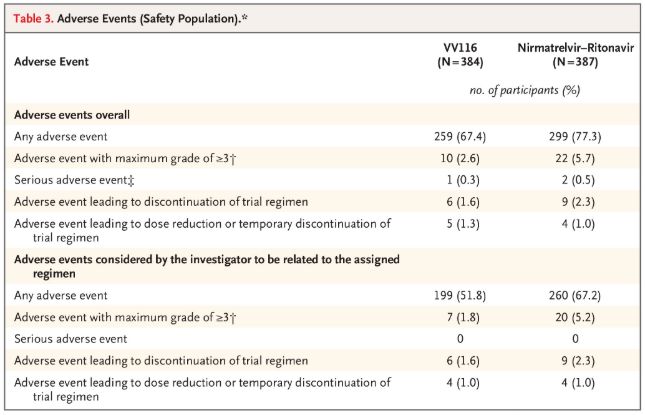
Uønskede hændelser (sikre personer)
Billedkilde: Reference 2
Kontroverser og spørgsmål
Den 23. maj 2022 offentliggjorde Juniper, at det kliniske fase III-registreringsstudie af VV116 versus PAXLOVID til tidlig behandling af mild til moderat COVID-19 (NCT05341609) nåede sit primære endepunkt.

Billedkilde: Reference 1
På et tidspunkt, hvor detaljer om forsøget manglede, var kontroversen omkring fase III-studiet dobbelt: for det første var det et enkeltblindet studie, og i mangel af en placebokontrol frygtede man, at det ville være vanskeligt at bedømme lægemidlet fuldstændig objektivt; for det andet var der spørgsmål om de kliniske endepunkter.
De kliniske inklusionskriterier for Juniper er (i) positive resultater for den nye kronetest, (ii) et eller flere milde eller moderate COVID-19-symptomer, og (iii) patienter med høj risiko for alvorlig COVID-19, herunder død. Det eneste primære kliniske endepunkt er dog 'tid til vedvarende klinisk bedring'.
Lige før annonceringen, den 14. maj, havde Juniper revideret de kliniske endepunkter ved at fjerne et af de kliniske primære endepunkter, "andel af konverteringer til alvorlig sygdom eller død" [3].
![]()
Billedkilde: Reference 1
Disse to hovedstridspunkter blev også specifikt behandlet i den offentliggjorte undersøgelse.
På grund af det pludselige udbrud af Omicron var produktionen af placebotabletter til Paxlovid ikke afsluttet før forsøgets start, og derfor var forskerne ikke i stand til at udføre dette forsøg ved hjælp af et dobbeltblindet, dobbelt-mock-design. Hvad angår det enkeltblindede aspekt af det kliniske forsøg, sagde Juniper, at protokollen blev udført efter kommunikation med regulerende myndigheder, og at det enkeltblindede design betyder, at hverken forskerne (inklusive evaluatoren af studiets endepunkt) eller sponsoren vil kende den specifikke terapeutiske lægemiddelallokering, før den endelige database er låst ved studiets afslutning.
Indtil tidspunktet for den endelige analyse havde ingen af deltagerne i forsøget oplevet død eller progression til en alvorlig Covid-19-hændelse, så der kan ikke drages nogen konklusioner om effekten af VV116 i forebyggelsen af progression til alvorlig eller kritisk Covid-19 eller død. Dataene indikerede, at den estimerede mediane tid fra randomisering til vedvarende regression af Covid-19-relaterede målsymptomer var 7 dage (95% CI, 7 til 8) i begge grupper (hazard ratio, 1,06; 95% CI, 0,91 til 1,22) [2]. Det er ikke vanskeligt at forklare, hvorfor det primære endepunkt 'konverteringsrate til alvorlig sygdom eller død', som oprindeligt blev sat før forsøgets afslutning, blev fjernet.
Den 18. maj 2022 offentliggjorde tidsskriftet Emerging Microbes & Infections resultaterne af det første kliniske forsøg med VV116 hos patienter inficeret med Omicron-varianten [4], et åbent, prospektivt kohortestudie med 136 bekræftede indlagte patienter.
Data fra studiet viste, at patienter med Omicron-infektion, der anvendte VV116 inden for 5 dage efter deres første positive nukleinsyretest, havde en tid til nukleinsyreregression på 8,56 dage, hvilket er mindre end de 11,13 dage i kontrolgruppen. Administration af VV116 til symptomatiske patienter inden for studiets tidsramme (2-10 dage efter den første positive nukleinsyretest) reducerede tiden til nukleinsyreregression hos alle patienter. Med hensyn til lægemiddelsikkerhed blev der ikke observeret alvorlige bivirkninger i VV116-behandlingsgruppen.
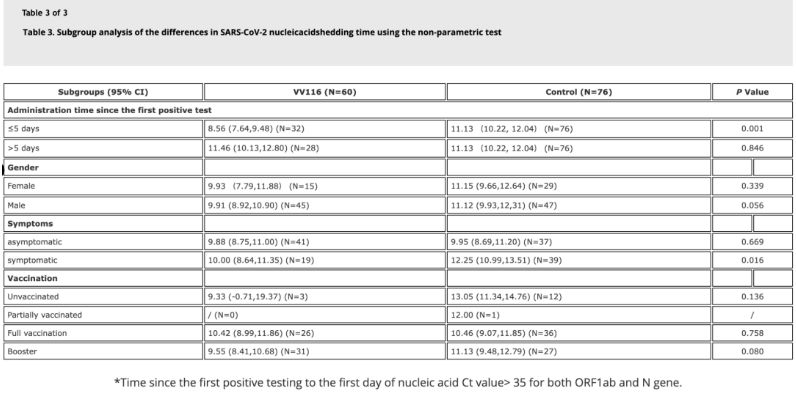
Billedkilde: Reference 4
Der er tre igangværende kliniske forsøg med VV116, hvoraf to er fase III-studier af mild til moderat COVID-19 (NCT05242042, NCT05582629). Det andet forsøg for moderat til svær COVID-19 er et internationalt multicenter, randomiseret, dobbeltblindet fase III klinisk studie (NCT05279235), der har til formål at evaluere effekten og sikkerheden af VV116 sammenlignet med standardbehandling. Ifølge Junipers meddelelse blev den første patient inkluderet og fik doser i marts 2022.

Billedkilde: clinicaltrials.gov
Referencer:
[1]Junshi Biotech: Meddelelse om det primære effektmål for fase III-registreret klinisk studie af VV116 versus PAXLOVID til tidlig behandling af mild til moderat COVID-19
[2]https: Yin, Zhiren Fu, Hao Xing, Li Li, Liying Sun, Heyu Huang, Quanbao Zhang, Linlin Xu, Yanting Jin, Rui Chen, Guoyue Lv, Zhijun Zhu, Wenhong Zhang, Zhengxin Wang. (2022) Omicron-infektionsprofil og vaccinationsstatus blandt 1881 levertransplantationsmodtagere: en multicenter retrospektiv kohorte. Emerging Microbes & Infections 11:1, side 2636-2644.
Opslagstidspunkt: 6. januar 2023
 Privatlivsindstillinger
Privatlivsindstillinger